Looking Back at the News on Florida Biotechnology News
Circa 2012 -2014
For a number of years this was the Florida Biotechnology News website where interested people could learn more about the Biotechnology news and developments from Florida. In addition to Florida biotech news the site also offered a directory of Florida biotech companies.
Content is from the site's 2014 archived pages providing a glimpse of what this site offered its readership.
FYI: Florida Biotechnology News Facebook posts ceased in 2017.
An observation: My parents retired to The Villages, an active adult community in Florida in 1995. When my mother died from complications of myeloma, my father chose to continue living there until his health deteriorated enough that I felt it was time to move him to the Hart Heritage Assisted Living facilities in Harford County, Maryland near to where I live. I looked at several homes for seniors, but finally decided on Hart Heritage Estates with their long term residential elder care facility in Forest Hills. Nestled on 6.5 acres of park-like grounds, the residents can enjoy nature in every season which I knew my father would enjoy. As long as my father can still walk, we plan to explore the nearby Ma and Pa trail, a 6.25-mile multi-purpose rail trail that follows the path of the old Ma and Pa Railroad through Harford County. My father avidly read the Florida Biotechnology News online while he was in Florida, particularly after my mother was diagnosed with myeloma cancer. Now that this site is no longer active, I have helped my father find other online sites with similar content. I'm pleased he still is motivated to get online, but I can start to see a slow but steady decline mentally, which instigated the move to Hart Heritage Estates.
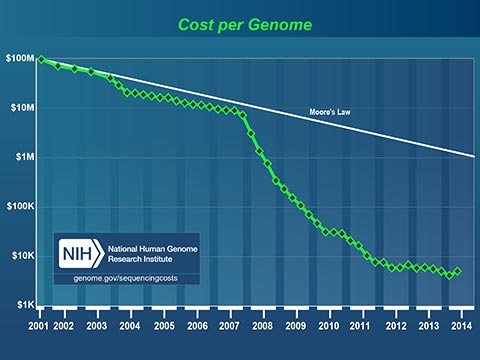
The plummeting cost to read a human genome will profoundly affect medicine in our lifetime. Today, I can spit in a test tube, mail it off with a check for $199 and receive a partial read of my DNA. I can learn my ancestry, inheritable conditions and even certain drug sensitivities. It now costs less to get a paternity test that it costs to hire a lawyer to fight about it!
Medicine has always been based on scientific studies of large groups of people. Medical practices and drugs are created to perform well for most of the people, most of the time. Fast, cheap genetic information is changing this model. In the near future, your personal genetic information will guide your personal medical care.
Since the mid 1990s, Florida has become home to more and more researchers and organizations working on the cutting edge of scientific investigation and discovery. They are working on what your genetic information means. They are learning how to collect and analyze this genetic information. In addition, they are learning how to use this new knowledge and information on everything from medical care and disease prevention to using algae and yeast to create fuel from sunlight or garbage.
This site is determined to keep Florida’s researchers up-to-date on what their co-workers, friends and rivals are up to across the state. We keep you current with developments in Tallahassee, the Research Coast and Lake Nona, and serve as a comprehensive listing of Florida’s Biotechnology companies. Finally, we toss in some interesting news from life science labs around the world, just to keep the ideas flowing. We hope you find something interesting, and when you do, please share your thoughts, insight and opinion.
Florida Bio Database contains profiles of biotechnology and biomedical device companies in Florida.
- Florida is home to nearly 40,000 Life Sciences establishments that employ about 691,000 people.
- Florida’s Life Sciences companies have a total payroll of more than $34.6 billion
![]()
2014 POSTS
Cancer stem cells identified in human patients
May 16, 2014
The gene mutations driving cancer have been tracked for the first time in patients back to a distinct set of cells at the root of cancer – cancer stem cells. The international research team, led by scientists at Karolinska Institutet and University of Oxford, studied a group of patients with myelodysplastic syndromes, a malignant blood condition which frequently develops into acute myeloid leukaemia.
The international research team, led by scientists at the University of Oxford and the Karolinska Institutet in Sweden, studied a group of patients with myelodysplastic syndromes – a malignant blood condition which frequently develops into acute myeloid leukaemia.
The researchers say their findings, Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo, reported in the journal Cancer Cell, offer conclusive evidence for the existence of cancer stem cells.
The concept of cancer stem cells has been a compelling but controversial idea for many years. It suggests that at the root of any cancer there is a small subset of cancer cells that are solely responsible for driving the growth and evolution of a patient’s cancer. These cancer stem cells replenish themselves and produce the other types of cancer cells, as normal stem cells produce other normal tissues.
The concept is important, because it suggests that only by developing treatments that get rid of the cancer stem cells will you be able to eradicate the cancer. Likewise, if you could selectively eliminate these cancer stem cells, the other remaining cancer cells would not be able to sustain the cancer.
‘It’s like having dandelions in your lawn. You can pull out as many as you want, but if you don’t get the roots they’ll come back,’ explains first author Dr Petter Woll of the MRC Weatherall Institute for Molecular Medicine at the University of Oxford.
The researchers, led by Professor Sten Eirik W Jacobsen at the MRC Molecular Haematology Unit and the Weatherall Institute for Molecular Medicine at the University of Oxford, investigated malignant cells in the bone marrow of patients with myelodysplastic syndrome (MDS) and followed them over time.
Using genetic tools to establish in which cells cancer-driving mutations originated and then propagated into other cancer cells, they demonstrated that a distinct and rare subset of MDS cells showed all the hallmarks of cancer stem cells, and that no other malignant MDS cells were able to propagate the tumour.
The MDS stem cells were rare, sat at the top of a hierarchy of MDS cells, could sustain themselves, replenish the other MDS cells, and were the origin of all stable DNA changes and mutations that drove the progression of the disease.
‘This is conclusive evidence for the existence of cancer stem cells in myelodysplastic syndromes,’ says Dr Woll. ‘We have identified a subset of cancer cells, shown that these rare cells are invariably the cells in which the cancer originates, and also are the only cancer-propagating cells in the patients. It is a vitally important step because it suggests that if you want to cure patients, you would need to target and remove these cells at the root of the cancer – but that would be sufficient, that would do it.’
The existence of cancer stem cells has already been reported in a number of human cancers, explains Professor Jacobsen, but previous findings have remained controversial since the lab tests used to establish the identity of cancer stem cells have been shown to be unreliable and, in any case, do not reflect the “real situation” in an intact tumour in a patient.
‘In our studies we avoided the problem of unreliable lab tests by tracking the origin and development of cancer-driving mutations in MDS patients,’ says Professor Jacobsen, who also holds a guest professorship at the Karolinska Institutet.
‘We need to understand more about what makes these cancer stem cells unique, what makes them different to all the other cancer cells. If we can find biological pathways that are specifically dysregulated in cancer stem cells, we might be able to target them with new drugs.’
Dr Woll cautions: ‘It is important to emphasize that our studies only investigated cancer stem cells in MDS, and that the identity, number and function of stem cells in other cancers are likely to differ from that of MDS.’

~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Mayo Clinic shows massive dose of Measles vaccine destroys cancer
May 14, 2014
In a proof of principle clinical trial, Mayo Clinic researchers have demonstrated that virotherapy — destroying cancer with a virus that infects and kills cancer cells but spares normal tissues — can be effective against the deadly cancer multiple myeloma. The findings appear in the journal Mayo Clinic Proceedings.
Two patients in the study received a single intravenous dose of an engineered measles virus (MV-NIS) that is selectively toxic to myeloma plasma cells. Both patients responded, showing reduction of both bone marrow cancer and myeloma protein. One patient, a 49-year-old woman, experienced complete remission of myeloma and has been clear of the disease for over six months.
“This is the first study to establish the feasibility of systemic oncolytic virotherapy for disseminated cancer,” says Stephen Russell, M.D., Ph.D., Mayo Clinic hematologist, first author of the paper and co-developer of the therapy. “These patients were not responsive to other therapies and had experienced several recurrences of their disease.”
Multiple myeloma is a cancer of plasma cells in the bone marrow, which also causes skeletal or soft tissue tumors. This cancer usually responds to immune system-stimulating drugs, but eventually overcomes them and is rarely cured.
In their article, the researchers explain they were reporting on these two patients because they were the first two studied at the highest possible dose, had limited previous exposure to measles, and therefore fewer antibodies to the virus, and essentially had no remaining treatment options.
Oncolytic virotherapy – using re-engineered viruses to fight cancer – has a history dating back to the 1950s. Thousands of cancer patients have been treated with oncolytic viruses from many different virus families (herpesviruses, poxviruses, common cold viruses, etc.). However, this study provides the first well-documented case of a patient with disseminated cancer having a complete remission at all disease sites after virus administration.
The second patient in the paper, whose cancer did not respond as well to the virus treatment, was equally remarkable because her imaging studies provided a clear proof that the intravenously administered virus specifically targeted the sites of tumor growth. This was done using high-tech imaging studies, which were possible only because the virus had been engineered with a ‘snitch gene’ — an easily identifiable marker — so researchers could accurately determine its location in the body.
More of the MV-NIS therapy is being manufactured for a larger, phase 2 clinical trial. The researchers also want to test the effectiveness of the virotherapy in combination with radioactive therapy (iodine-131) in a future study.
Other authors include Mark Federspiel, Ph.D., Kah-Whye Peng, Ph.D., M.Med., Caili Tong, David Dingli, M.D., Ph.D., William Morice, M.D., Ph.D., Val Lowe, M.D., Michael O’Connor, Ph.D., Robert Kyle, M.D., Nelson Leung, M.D., Francis Buadi, M.D., S. Vincent Rajkumar, M.D., Morie Gertz, M.D., Martha Lacy, M.D., and senior and corresponding author Angela Dispenzieri, M.D., all of Mayo Clinic.
The research was supported by the National Institutes of Health-National Cancer Institute, Al and Mary Agnes McQuinn, The Harold W. Siebens Foundation; and The Richard M. Schulze Family Foundation.
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
First Living Organism that Transmits Added Letters in DNA ‘Alphabet’
May 8, 2014
Scientists at The Scripps Research Institute (TSRI) in La Jolla, California have engineered a bacterium whose genetic material includes an added pair of DNA “letters,” or bases, not found in nature. The cells of this unique bacterium can replicate the unnatural DNA bases more or less normally, for as long as the molecular building blocks are supplied. “A semi-synthetic organism with an expanded genetic alphabet” appears in the journal Nature.
“Life on Earth in all its diversity is encoded by only two pairs of DNA bases, A-T and C-G, and what we’ve made is an organism that stably contains those two plus a third, unnatural pair of bases,” said TSRI Associate Professor Floyd E. Romesberg, who led the research team. “This shows that other solutions to storing information are possible and, of course, takes us closer to an expanded-DNA biology that will have many exciting applications—from new medicines to new kinds of nanotechnology.”
Many Challenges
Romesberg and his laboratory have been working since the late 1990s to find pairs of molecules that could serve as new, functional DNA bases—and, in principle, could code for proteins and organisms that have never existed before.
The task hasn’t been a simple one. Any functional new pair of DNA bases would have to bind with an affinity comparable to that of the natural nucleoside base-pairs adenine–thymine and cytosine–guanine. Such new bases also would have to line up stably alongside the natural bases in a zipper-like stretch of DNA. They would be required to unzip and re-zip smoothly when worked on by natural polymerase enzymes during DNA replication and transcription into RNA. And somehow these nucleoside interlopers would have to avoid being attacked and removed by natural DNA-repair mechanisms.
Despite these challenges, by 2008 Romesberg and his colleagues had taken a big step towards this goal; in a study published that year, they identified sets of nucleoside molecules that can hook up across a double-strand of DNA almost as snugly as natural base pairs and showed that DNA containing these unnatural base pairs can replicate in the presence of the right enzymes. In a study that came out the following year, the researchers were able to find enzymes that transcribe this semi-synthetic DNA into RNA.
But this work was conducted in the simplified milieu of a test tube. “These unnatural base pairs have worked beautifully in vitro, but the big challenge has been to get them working in the much more complex environment of a living cell,” said Denis A. Malyshev, a member of the Romesberg laboratory who was lead author of the new report.
Microalgae Lead to Breakthrough
In the new study, the team synthesized a stretch of circular DNA known as a plasmid and inserted it into cells of the common bacterium E. coli. The plasmid DNA contained natural T-A and C-G base pairs along with the best-performing unnatural base pair Romesberg’s laboratory had discovered, two molecules known as d5SICS and dNaM. The goal was to get the E. coli cells to replicate this semi-synthetic DNA as normally as possible.
The greatest hurdle may be reassuring to those who fear the uncontrolled release of a new life form: the molecular building blocks for d5SICS and dNaM are not naturally in cells. Thus, to get the E. coli to replicate the DNA containing these unnatural bases, the researchers had to supply the molecular building blocks artificially, by adding them to the fluid solution outside the cell. Then, to get the building blocks, known as nucleoside triphosphates, into the cells, they had to find special triphosphate transporter molecules that would do the job.
The researchers eventually were able to find a triphosphate transporter, made by a species of microalgae, that was good enough at importing the unnatural triphosphates. “That was a big breakthrough for us—an enabling breakthrough,” said Malyshev.
Though the completion of the project took another year, no hurdles that large arose again. The team found, somewhat to their surprise, that the semi-synthetic plasmid replicated with reasonable speed and accuracy, did not greatly hamper the growth of the E. coli cells, and showed no sign of losing its unnatural base pairs to DNA repair mechanisms.
“When we stopped the flow of the unnatural triphosphate building blocks into the cells, the replacement of d5SICS–dNaM with natural base pairs was very nicely correlated with the cell replication itself—there didn’t seem to be other factors excising the unnatural base pairs from the DNA,” Malyshev said. “An important thing to note is that these two breakthroughs also provide control over the system. Our new bases can only get into the cell if we turn on the ‘base transporter’ protein. Without this transporter or when new bases are not provided, the cell will revert back to A, T, G, C, and the d5SICS and dNaM will disappear from the genome.”
The next step will be to demonstrate the in-cell transcription of the new, expanded-alphabet DNA into the RNA that feeds the protein-making machinery of cells. “In principle, we could encode new proteins made from new, unnatural amino acids—which would give us greater power than ever to tailor protein therapeutics and diagnostics and laboratory reagents to have desired functions,” Romesberg said. “Other applications, such as nanomaterials, are also possible.”
Other contributors to the paper, “A semi-synthetic organism with an expanded genetic alphabet,” were Kirandeep Dhami, Thomas Lavergne and Tingjian Chen of TSRI, and Nan Dai, Jeremy M. Foster and Ivan R. Corrêa Jr. of New England Biolabs, Inc.
The research was funded in part by the US National Institutes of Health (GM 060005).
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
UF researchers create protein with potential for weight loss, diabetes treatment
May 2, 2014
It sounds like a magic bullet: Administer a protein, watch the subject lose weight.
But that’s exactly what University of Florida scientists found when they discovered a new way to deliver a protein that helps develop cells that convert fat into energy.
The study builds upon on a discovery by Bruce Spiegelman, a cell biologist at the Boston-based Dana-Farber Cancer Institute, who found that human muscles release a hormone he called irisin during exercise. Spiegelman also found that mice lost a small amount of weight when given the irisin gene using a virus to ferry it into cells.
Now the UF team — including researcher Dr. Li-Jun Yang, Shi-Wu Li, and postdoctoral researcher William Donelan — has for the first time created a stable protein form of irisin, opening the door to human studies that weren’t previously possible because the virus has not been approved for use in people.
“We found that if you just deliver irisin protein to obese mice, the mice can actually reduce body weight without exercise. This study may have important implications for the therapy of obesity and Type 2 diabetes,” Yang said.
Li, a research associate in the UF department of immunology, pathology and laboratory medicine, spearheaded the research. In collaboration with a group of scientists including Yuan Zhang, a graduate student in the Center for Stem Cell and Regenerative Medicine in China, Li conducted the trials in mice. The mice were fed a high-fat diet for 10 weeks, and then divided into two groups. One group was injected with irisin daily for two weeks while the other group was given saline daily for two weeks. The control group gained an average of 1.2 percent of body weight over the two-week period. The group treated with irisin lost a significant amount of weight, about 2 percent of the mice’s body weight. The group was also more sensitive to insulin, which helps them store food as energy, possibly benefitting people with Type 2 diabetes.
The body contains two types of fat: white fat cells and brown fat cells. White fat cells store fat, and brown fat cells convert fat into energy.
During exercise, muscles receive stress signals that trigger muscle cells to release irisin. The irisin travels to white fat tissues, where researchers think it binds to receptors on the fat cells, stimulating the transformation of white fat cells into energy-burning brown fat cells.
As a result, more fat can be burned, UF researchers said.
“The ancient Greek goddess Iris was the messenger of the gods,” Donelan said. “Irisin is a sort of messenger molecule, in a sense. It changes exercise into positive results for the organism.”
Aside from spurring weight loss, the hormone also may help treat a host of other metabolic disorders, Donelan said.
“You don’t want to tell people to not exercise, but if you did get people to exercise and use irisin, it could be a pretty good treatment for conditions such as Type 2 diabetes, obesity or any type of metabolic disorders,” he said.
The team published its findings in the February volume of the journal Diabetes. The protein is nearly identical to natural human irisin, so it could be used instead of the viral vector, said Yang. Spiegelman agreed, calling the researchers’ work a “substantial step in the direction of human therapeutics,” in a review of the researchers’ study published in the same issue of Diabetes.
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Scientists regenerate immune organ in mice
April 9, 2014
Scientists have for the first time used regenerative medicine to fully restore a degenerated organ in a living animal, a discovery that could pave the way for future human therapies.
The team from the Medical Research Council (MRC) Centre for Regenerative Medicine, at the University of Edinburgh, rebuilt the thymus of very old mice by reactivating a natural mechanism that gets shut down with age.
The regenerated thymus was very similar to one in a young mouse in terms of structure and the genes expressed. The function of the organ was also restored, and mice receiving the treatment began making more T cells – a type of white blood cell important in fighting infection. However, the researchers do not yet know if the immune system of the older mice was strengthened. The research is published today in the journal Development.
Professor Clare Blackburn from the MRC Centre for Regenerative Medicine, at the University of Edinburgh, who led the research, said: “By targeting a single protein, we have been able to almost completely reverse age-related shrinking of the thymus. Our results suggest that targeting the same pathway in humans may improve thymus function and therefore boost immunity in elderly patients, or those with a suppressed immune system. However, before we test this in humans we need to carry out more work to make sure the process can be tightly controlled.”
The thymus, located in front of the heart, is the first organ to deteriorate as we age. This shrinking is one of the main reasons our immune system becomes less effective and we lose the ability to fight off new infections, such as flu, as we get older.
Researchers targeted a key part of this process – a protein called FOXN1, which helps to control how important genes in the thymus are switched on. They used genetically modified mice to enable them to increase levels of this protein using chemical signals. By doing so they managed to instruct immature cells in the thymus – similar to stem cells – to rebuild the organ in the older mice. The regenerated thymus was more than twice the size than in the untreated mice.
Dr Rob Buckle, Head of Regenerative Medicine at the MRC, said: “One of the key goals in regenerative medicine is harnessing the body’s own repair mechanisms and manipulating these in a controlled way to treat disease. This interesting study suggests that organ regeneration in a mammal can be directed by manipulation of a single protein, which is likely to have broad implications for other areas of regenerative biology.”
Previous attempts to provoke thymus regeneration have involved using sex hormones, but these have resulted in only temporary recovery of size and function of the organ. In this study, the recovery of the thymus was sustainable, but more work is needed to ensure there are no unintended consequences of increasing FOXN1.
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Lab-grown muscle is the first to heal itself after animal implantation
April 8, 2014
Biomedical engineers have grown living skeletal muscle that looks a lot like the real thing. It contracts powerfully and rapidly, integrates into mice quickly, and for the first time, demonstrates the ability to heal itself both inside the laboratory and inside an animal.
The study conducted at Duke University tested the bioengineered muscle by literally watching it through a window on the back of living mouse. The novel technique allowed for real-time monitoring of the muscle’s integration and maturation inside a living, walking animal.
Both the lab-grown muscle and experimental techniques are important steps toward growing viable muscle for studying diseases and treating injuries, said Nenad Bursac, associate professor of biomedical engineering at Duke. Biomimetic engineered muscle with capacity for vascular integration and functional maturation in vivo appears in the Proceedings of the National Academy of Sciences Early Edition.
“The muscle we have made represents an important advance for the field,” Bursac said. “It’s the first time engineered muscle has been created that contracts as strongly as native neonatal skeletal muscle.”
Through years of perfecting their techniques, a team led by Bursac and graduate student Mark Juhas discovered that preparing better muscle requires two things—well-developed contractile muscle fibers and a pool of muscle stem cells, known as satellite cells.
Every muscle has satellite cells on reserve, ready to activate upon injury and begin the regeneration process. The key to the team’s success was successfully creating the microenvironments—called niches—where these stem cells await their call to duty.
“Simply implanting satellite cells or less-developed muscle doesn’t work as well,” said Juhas. “The well-developed muscle we made provides niches for satellite cells to live in, and, when needed, to restore the robust musculature and its function.”
To put their muscle to the test, the engineers ran it through a gauntlet of trials in the laboratory. By stimulating it with electric pulses, they measured its contractile strength, showing that it was more than 10 times stronger than any previous engineered muscles. They damaged it with a toxin found in snake venom to prove that the satellite cells could activate, multiply and successfully heal the injured muscle fibers.
Then they moved it out of a dish and into a mouse.
With the help of Greg Palmer, an assistant professor of radiation oncology in the Duke University School of Medicine, the team inserted their lab-grown muscle into a small chamber placed on the backs of live mice. The chamber was then covered by a glass panel. Every two days for two weeks, Juhas imaged the implanted muscles through the window to check on their progress.
By genetically modifying the muscle fibers to produce fluorescent flashes during calcium spikes—which cause muscle to contract— the researchers could watch the flashes become brighter as the muscle grew stronger.
“We could see and measure in real time how blood vessels grew into the implanted muscle fibers, maturing toward equaling the strength of its native counterpart,” said Juhas.
The engineers are now beginning work to see if their biomimetic muscle can be used to repair actual muscle injuries and disease.
“Can it vascularize, innervate and repair the damaged muscle’s function?” asked Bursac. “That is what we will be working on for the next several years.”
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Tobacco plant’s natural defense promising against cancer cells in vitro
April 7, 2014
Scientists at the La Trobe Institute for Molecular Science have identified that a molecule occurring in the flower of the tobacco plant that fights off fungi and bacteria also has the ability to identify and destroy cancer. Phosphoinositide-mediated oligomerization of a defensin induces cell lysis
The defence molecule, called NaD1, works by forming a pincer-like structure that grips onto lipids present in the membrane of cancer cells and rips it open, causing the cell to expel its contents and explode.
Lead investigator Dr Mark Hulett said the discovery has potential for use in cancer treatment.
‘There is some irony in the fact that a powerful defence mechanism against cancer is found in the flower of a species of ornamental tobacco plant, but this is a welcome discovery, whatever the origin,’ Dr Hulett said.
‘The next step is to undertake pre-clinical studies to determine what role NaD1 might be able to play in treating cancer. The preclinical work is being conducted by the Melbourne biotechnology company Hexima. So far the preliminary trials have looked promising.
‘We are confident there is potential for this discovery to translate to therapeutic use in humans.
‘One of the biggest issues with current cancer therapies is that the effect of the treatment is indiscriminate,’ Dr Hulett said. ‘In contrast, we’ve found NaD1 can target cancerous cells and has little or no effect on those that are healthy.’
Dr Hulett said scientists have known for some time about the molecules that form the first line of defence against microbial invasion in all plant and animal species. Until now nobody has known how the molecules actually did their job.
‘We’ve discovered the workings of this universal defence process, which could also potentially be harnessed for the development of other therapeutic applications, including antibiotic treatment for microbial infections,’ he said.
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Scripps Florida Scientists Devise New, Lower Cost Method to Create More Usable Fuels
March 14, 2014
As the United States continues to lead the world in the production of natural gas, scientists from the Florida campus of The Scripps Research Institute (TSRI) have devised a new and more efficient method with the potential to convert the major components found in natural gas into useable fuels and chemicals—opening the door to cheaper, more abundant energy and materials with much lower emissions.
The research, which was led by TSRI Professor Roy Periana, uses clever chemistry and nontraditional materials to turn natural gas into liquid products at much lower temperatures than conventional methods.
“We uncovered a whole new class of inexpensive metals that allows us to process methane and the other alkanes contained in natural gas, ethane and propane, at about 180 degrees centigrade or lower, instead of the more than 500oC used in current processes,” said Periana. “This creates the potential to produce fuels and chemicals at an extraordinarily lower cost.”
The research was described in the March 14, 2014 edition of the journalScience.
The Challenge
Methane is the most abundant compound in natural gas. However, converting methane into a useable, versatile liquid product remains a costly and complicated process that has changed little from the original process developed in the 1940s. But with the boom in natural gas discovery growing every day, new processes are needed to convert methane to fuel and chemicals that can compete economically with production from petroleum.
Methane, ethane and propane, the major components in natural gas, belong to a class of molecules named alkanes that are the simplest hydrocarbons and one of the most abundant, cleanest sources of energy and materials. However, transportation can be expensive and converting these alkanes into other useful forms such as gasoline, alcohols or olefins is expensive and often inefficient.
At the core of technologies for converting the alkanes in natural gas is the chemistry of the carbon-hydrogen. Because of the high strength of these bonds, current processes for converting these alkanes employ high temperatures (more than 500oC) that lead to high costs, high emissions and lower efficiencies.
The development of lower temperature (less than 250oC), selective, alkane carbon-hydrogen bond conversion chemistry could lead to a major shift in energy and materials production technology.
An Elegant Solution
Periana has been thinking about this type of problem for decades in pursuit of lower-cost, environmentally friendly energy solutions and has designed some of the most efficient systems (Periana et. al., Science 1993, 1998 and 2003) for alkane conversion that operate at lower temperatures.
However, when Periana and his team examined these first-generation systems they realized that the precious metals they used, such as platinum, palladium, rhodium, gold, were both too expensive and rare for widespread use.
“What we wanted were elements that are more abundant and much less expensive that can carry out the same chemistry under more practical conditions,” said Brian G. Hashiguchi, the first author of the study and a member of Periana’s lab. “We also wanted to find materials that could convert methane as well as the other major components in natural gas, ethane and propane.”
Approaching the problem both theoretically and experimentally, the team hit on inexpensive metals known as main group elements, some of which are byproducts of refining certain ores. For example, one of the materials can be made from common lead dioxide, a synthetic compound used in the production of matches and fireworks.
“The reaction of alkanes with this class of materials we’ve identified is novel,” Periana said. “They can react with methane, ethane as well as propane at lower temperatures with extraordinarily selectivity—and produce the corresponding alcohols as the only the desired products. These products are all major commodity chemicals and are also ideal, inexpensive sources for fuels and plastics.”
If successfully developed, new process using these metals could potentially allow the large reserves of natural gas in the United States to be used as alternative resources for fuels and chemicals.
“This is a highly novel piece of work that opens the way to upgrading of natural gas to useful chemicals with simple materials and moderate conditions,” said Robert Crabtree, a chemistry professor at Yale who is familiar with the new study.
In addition to Periana and Hashiguchi, authors of the study, “Main-Group Compounds Selectively Oxidize Mixtures of Methane, Ethane, and Propane to Alcohol Esters,” are Michael M. Konnick, Steven M. Bischof and Niles Gunsalus of The Scripps Research Institute; and Samantha J. Gustafson, Deepa Devarajan and Daniel H. Ess of Brigham Young University.
This study was supported by the U.S. Department of Energy (DE-SC0001298).
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
![]()
Medical Device Companies
Due to popular demand, we’ve got a Florida “Medical Device” manufacturer list coming together… Please add more names, corrections or deletions in the comments. Thanks
- Baxter
- Beckman Coulter
- Benz Research & Development
- Biomet 3i
- Bolton Medical Solutions
- Bovie Medical
- Clavina Diagnostics
- Command Medical Products
- Cordis
- Dayamed
- Emunamedica
- eTect
- Exactech
- GeNO
- HydroMAR
- HyGreen
- International Medical Industries
- Innovia
- Iradimed
- Johnson & Johnson Vistakon in Jacksonville
- KeriCure
- Mako Surgical
- Medtronic Surgical Technologies
- Miami Cardiovascular Innovations
- Nims
- OrbusNeich
- Orthosensor
- Rapid Pathogen Screening
- RTI Biologics
- Smith & Nephew
- Xhale
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Basic Timeline of DNA investigation
• 1953 Discovery of the structure of the DNA double helix.
• 1972 Development of recombinant DNA technology, which permits isolation of defined fragments of DNA; prior to this, the only accessible samples for sequencing were from bacteriophage or virus DNA.
• 1977 The first complete DNA genome to be sequenced is that of bacteriophage fX174.
• 1977 Allan Maxam and Walter Gilbert publish “DNA sequencing by chemical degradation”. Frederick Sanger, independently, publishes “DNA sequencing with chain-terminating inhibitors”.
• 1984 Medical Research Council scientists decipher the complete DNA sequence of the Epstein-Barr virus, 170 kb.
• 1986 Leroy E. Hood’s laboratory at the California Institute of Technology and Smith announce the first semi-automated DNA sequencing machine.
• 1987 Applied Biosystems markets first automated sequencing machine, the model ABI 370.
• 1991 Sequencing of human expressed sequence tags begins in Craig Venter’s lab, an attempt to capture the coding fraction of the human genome.
• 1995 Craig Venter, Hamilton Smith, and colleagues at The Institute for Genomic Research (TIGR) publish the first complete genome of a free-living organism, the bacterium Haemophilus influenzae. The circular chromosome contains 1,830,137 bases and its publication in the journal Science marks the first use of whole-genome shotgun sequencing, eliminating the need for initial mapping efforts.
• 2000 Lynx Therapeutics publishes and markets “MPSS” – a parallelised, adapter/ligation-mediated, bead-based sequencing technology, launching “next-generation” sequencing.
• 2001 A draft sequence of the human genome is published.